
Command Line Options#
After installing StACKER, the command stacker is added
to terminal. Different routines can be run in the command Line
using stacker -s ROUTINE. Running stacker --help
will give the subroutine options:
[user]$ stacker --help
usage: stacker -s ROUTINE [-h]
Wrapper to run stacker subroutines using the -s flag.
More info on each routine given by `stacker -s ROUTINE -h`
options:
-s, --script ROUTINE Name of command to use. Options for ROUTINE:
filter_traj:
filters trajectory and topology files to desired residue numbers and atom names
bottaro OR pairwise OR psf:
Create Polar Stacking Fingerprint like those in Figure 1 of Bottaro et. al (https://doi.org/10.1093/nar/gku972)
res_distance:
Get the distance between two residues in a given frame
system OR ssf:
Create a System Stacking Fingerprint of distances by residue
stack_events:
Get list of residues with most stacking events (distance closest to 3.5Å)
compare:
Get the most changed stacking events between two fingerprints using the outputs of stacker -s stack_events
-h, --help show this help message and exit
Filtering Trajectory#
Running stacker -s filter_traj will filter a trajectory by
residue/atom indices and output the trajectory to a .pdb.
Running stacker -s filter_traj --help will give more flag options:
[user]$ stacker -s filter_traj --help
usage: stacker -s ROUTINE -trj TRAJECTORY_FILENAME -top TOPOLOGY_FILENAME -o OUTPUT_FILE [-r RESIDUES] [-a ATOM_NAMES] [-h]
Filters trajectory and topology files to desired residue numbers and atom names and outputs to a PDB
Examples:
[user]$ stacker -s filter_traj -trj testing/first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd -top testing/5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop -o testing/command_line_tests/filter/5JUP_N2_tUAG_aCUA_+1GCU_nowat_mdcrd.pdb -r 426,427 -a C2,C4,C6
options:
-s, --script ROUTINE Name of command to use. Options for ROUTINE:
filter_traj:
filters trajectory and topology files to desired residue numbers and atom names
bottaro OR pairwise OR psf:
Create Polar Stacking Fingerprint like those in Figure 1 of Bottaro et. al (https://doi.org/10.1093/nar/gku972)
res_distance:
Get the distance between two residues in a given frame
system OR ssf:
Create a System Stacking Fingerprint of distances by residue
stack_events:
Get list of residues with most stacking events (distance closest to 3.5Å)
compare:
Get the most changed stacking events between two fingerprints using the outputs of stacker -s stack_events
-r, --residues RESIDUES
Smart-indexed list of 1-indexed residues, also accepts dash (-) list creation (eg. 1-5,10 = 1,2,3,4,5,10)
-a, --atom_names ATOM_NAMES
Comma-separated list of atom names to filter
-h, --help show this help message and exit
Required Arguments:
-trj, --trajectory TRAJECTORY_FILENAME
Filepath to trajectory file for the MD simulation
-top, --topology TOPOLOGY_FILENAME
Filepath to Topology file for the MD simulation
-o, --output OUTPUT_FILE
Filepath of PDB to output to
This is the command line equivalent of filter_traj_to_pdb(). So, the following are equivalent:
>>> import stacker as st
>>> st.filter_traj_to_pdb(
... trj_file = "first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd",
... top_file = "5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop",
... pdb = "5JUP_N2_tUAG_aCUA_+1GCU_nowat_mdcrd.pdb",
... residues = "426,427",
... atoms = {"C2","C4","C6"}
... )
[user] $ stacker -s filter_traj \
-trj first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd \
-top 5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop \
-o 5JUP_N2_tUAG_aCUA_+1GCU_nowat_mdcrd.pdb \
-r 426,427 \
-a C2,C4,C6
Get Distance Between Residues#
Running stacker -s res_distance will give the distance in Angstroms
between the center of geometry of two residues for a given frame.
Running stacker -s res_distance --help will give more flag options:
[user]$ stacker -s res_distance --help
usage: stacker -s ROUTINE -trj TRAJECTORY_FILENAME -top TOPOLOGY_FILENAME [-f FRAME_NUM] -r RESIDUES [-b N_FRAMES] [-a ATOM_NAMES] [-h]
Get the distance between two residues in a given frame
Examples:
[user]$ stacker -s res_distance -trj testing/first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd -top testing/5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop -f 2 --residues 426,427 --atom_names C2,C4,C6
options:
-s, --script ROUTINE Name of command to use. Options for ROUTINE:
filter_traj:
filters trajectory and topology files to desired residue numbers and atom names
bottaro OR pairwise OR psf:
Create Polar Stacking Fingerprint like those in Figure 1 of Bottaro et. al (https://doi.org/10.1093/nar/gku972)
res_distance:
Get the distance between two residues in a given frame
system OR ssf:
Create a System Stacking Fingerprint of distances by residue
stack_events:
Get list of residues with most stacking events (distance closest to 3.5Å)
compare:
Get the most changed stacking events between two fingerprints using the outputs of stacker -s stack_events
-f, --frame FRAME_NUM
1-indexed Frame Number within trajectory to analyze
-b, --bootstrap N_FRAMES
Run bootstrap analysis on this residue pairing, sampling N_FRAMES with replacement
-a, --atom_names ATOM_NAMES
Comma-separated list of atom names. Three required to get center of geometry for a residue. default = C2,C4,C6
-h, --help show this help message and exit
Required Arguments:
-trj, --trajectory TRAJECTORY_FILENAME
Filepath to trajectory file for the MD simulation
-top, --topology TOPOLOGY_FILENAME
Filepath to Topology file for the MD simulation
-r, --residues RESIDUES
Smart-indexed list of 1-indexed residues, must provide only 2 residues, accepts dash (-) list creation (eg. 1-5,10 = 1,2,3,4,5,10)
This is the command line equivalent of get_residue_distance_for_frame().
So, the following are equivalent:
>>> import stacker as st
>>> filtered_traj = st.filter_traj('first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd',
... '5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop',
... residues = {426,427},
... atoms = {'C2','C4','C6'})
>>> st.get_residue_distance_for_frame(
... trj = filtered_traj,
... frame = 3,
... res1_atoms = ("C2", "C4", "C6"),
... res2_atoms = ("C2", "C4", "C6"),
... write_output = True
...)
[user] $ stacker -s res_distance \
-trj first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd \
-top 5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop \
-r 426,427 \
-f 3 \
-a C2,C4,C6
Create System Stacking Fingerprints (SSFs)#
Running stacker -s ssf will create a System Stacking
Fingerprint (SSF) for the trajectory, with many options to customize
the plot outputs. Running stacker -s ssf --help will give
more flag options:
[user]$ stacker -s ssf --help
usage: stacker -s ROUTINE -trj TRAJECTORY_FILENAME -top TOPOLOGY_FILENAME [-r RESIDUES] [-i INPUT_FILE] [-f FRAME_NUM | -fl FRAME_LIST] [-o OUTPUT_FILE]
[-g N_EVENTS] [-d OUTPUT_FILE] [-B INPUT_FILE] [-l LIMITS] [-y SCALE_STYLE] [-t N_THREADS] [-h]
Creates a System Stacking Fingerprint of the average structure across the chosen frames of a trajectory.
Examples:
[user]$ stacker -s system -trj testing/first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd -top testing/5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop -r 90-215 -fl 1-2
[user]$ stacker -s ssf -trj testing/first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd -top testing/5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop -r 90-215 -fl 1-2 -g 10 -o testing/command_line_tests/pairwise/5JUP_N2_tUAG_aCUA_+1GCU_nowat_pairwise_avg_1to2.png -d testing/command_line_tests/pairwise/5JUP_N2_tUAG_aCUA_+1GCU_data_1to2.txt
options:
-s, --script ROUTINE Name of command to use. Options for ROUTINE:
filter_traj:
filters trajectory and topology files to desired residue numbers and atom names
bottaro OR pairwise OR psf:
Create Polar Stacking Fingerprint like those in Figure 1 of Bottaro et. al (https://doi.org/10.1093/nar/gku972)
res_distance:
Get the distance between two residues in a given frame
system OR ssf:
Create a System Stacking Fingerprint of distances by residue
stack_events:
Get list of residues with most stacking events (distance closest to 3.5Å)
compare:
Get the most changed stacking events between two fingerprints using the outputs of stacker -s stack_events
-r, --residues RESIDUES
Smart-indexed list of 1-indexed residues, also accepts dash (-) list creation (eg. 1-5,10 = 1,2,3,4,5,10)
-i, --input INPUT_FILE
Input .txt file containing per-frame stacking information, in lieu of running stacking fingerprint analysis again.
TXT file can be created by running `stacker -s system -d OUTPUT_FILE`
-r flag must match the residues used to create the TXT file
-f, --frame FRAME_NUM
1-indexed Frame Number within trajectory to analyze, cannot be used with -fl
-fl, --frame_list FRAME_LIST
Smart-indexed list of 1-indexed Frame Numbers within trajectory to analyze,
gets average distance between residues across these frames
if empty all frames are used, cannot be used with -f
-o, --output OUTPUT_FILE
Filename of output PNG to write plot to. If empty, will output displays to Python visual
-g, --get_stacking N_EVENTS
Get list of N_EVENTS residues with most stacking events (distance closest to 3.5Å) in the average structure across all frames.
Print to standard output. Equivalent to -s stack_events -n N_EVENTS
-d, --data_output OUTPUT_FILE
Output the calculated per-frame numpy arrays that create the stacking fingerprint matrix to a file
-B, --input_B INPUT_FILE
Input .txt file containing per-frame stacking information for a second fingerprint, creates fingerprint where top left is initial input, bottom right is second fingerprint.
Used in lieu of running stacking fingerprint analysis again.
TXT file can be created by running `stacker -s system -d OUTPUT_FILE`
-r flag must match the residues used to create the TXT file
-l, --limits LIMITS limits of the color scale, default = (0,7)
-y, --scale_style SCALE_STYLE
style of color scale. {bellcurve, gradient}
-t, --threads N_THREADS
Use multithreading with INT worker threads
-h, --help show this help message and exit
Required Arguments:
-trj, --trajectory TRAJECTORY_FILENAME
Filepath to trajectory file for the MD simulation
-top, --topology TOPOLOGY_FILENAME
Filepath to Topology file for the MD simulation
Get Average SSF#
The basic SSF Command Line function runs get_frame_average() and
system_stacking_fingerprints(), creating an SSF for the
trajectory averaged across all frames:
>>> import stacker as st
>>> filtered_traj = st.filter_traj('first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd',
'5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop',
residues = '90-215',
atoms = {'C2','C4','C6'})
>>> ssfs = st.system_stacking_fingerprints(filtered_traj, frames = '1-2', write_output = False)
>>> avg_ssf = st.get_frame_average(ssfs)
>>> resSeqs = [res.resSeq for res in filtered_traj.topology.residues]
>>> st.display_ssfs(avg_ssf, resSeqs)
[user]$ stacker -s ssf \
-trj first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd \
-top 5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop \
-r 90-215 \
-fl 1-2
Data Output#
There are three important flags to output the SSF data:
-o, --output OUTPUT_FILE: Save SSF to an output PNGOUTPUT_FILE.-g, --get_stacking N_EVENTS: Prints top stacking pairs to standard output.
Equivalent to get_top_stacking(), where N_EVENTS is the n_events parameter.
- -d, --data_output OUTPUT_FILE : Output the list of SSFs to .txt.gz. You can input
this SSF data rather than recalculate each time.
Expanding on the command above, we create SSFs for frames 1-10 and use
-t 10 to calculate them in parallel:
[user]$ stacker -s ssf \
-trj first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd \
-top 5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop \
-r 90-215 \
-fl 1-10 \
-o 5JUP_N2_tUAG_aCUA_+1GCU_nowat.SSF.png \
-d 5JUP_N2_tUAG_aCUA_+1GCU_nowat.data.txt.gz \
-g 5 \
-t 10
Res1 Res2 Avg_Dist
117 108 3.43
127 94 3.67
93 130 3.68
177 204 3.80
197 195 3.85
This outputs the average SSF for frames 1-10 to 5JUP_N2_tUAG_aCUA_+1GCU_nowat.SSF.png:
The list of top stacking events can be customized much further via the
stacker -s stack_events command. See “Get Top Stacking Events” below.
The -d flag above outputs the SSF data for each frame to
5JUP_N2_tUAG_aCUA_+1GCU_nowat.data.txt.gz. We can read this into a
Python object representing the 10 frames of 126x126 residue SSFs
with load_ssfs():
>>> import stacker as st
>>> ssfs = st.load_ssfs("5JUP_N2_tUAG_aCUA_+1GCU_nowat.data.txt.gz")
>>> ssfs.shape
(10, 126, 126)
Inputting SSF Data#
In lieu of recalculating SSF data for a trajectory, the .txt.gz file
outputted by stacker -s ssf -d OUTFILE can be loaded in using:
-i, --input INPUT_FILE: A.txtor.txt.gzcontaining per-frame SSF
data created by stacker -s ssf -d OUTFILE.
- -B, --input_B INPUT_FILE : A second .txt or .txt.gz file that will be displayed
on the bottom left of the output SSF, allowing comparison between two SSFs. Must be the same
shape as the data inputted by the -i flag.
When using these, the -trj, -top, and -r flags should be the same as those used
to create either input. In the code below, we input two .txt.gz files representing SSF
data from the same structure with two different mRNA codons:
[user]$ stacker -s ssf \
-trj 5JUP_N2_tGGG_aCCU_+1GCU_nowat_2fpns.mdcrd \
-top 5JUP_N2_tGGG_aCCU_+1GCU_nowat.prmtop \
-r 2-5,13-16,23-31,46-51,65-76,88-104,122-141,164-175,184-198,288-289,401-415,420-430 \
-i 5JUP_N2_tGGG_aCCU_+1GCU_data.txt.gz
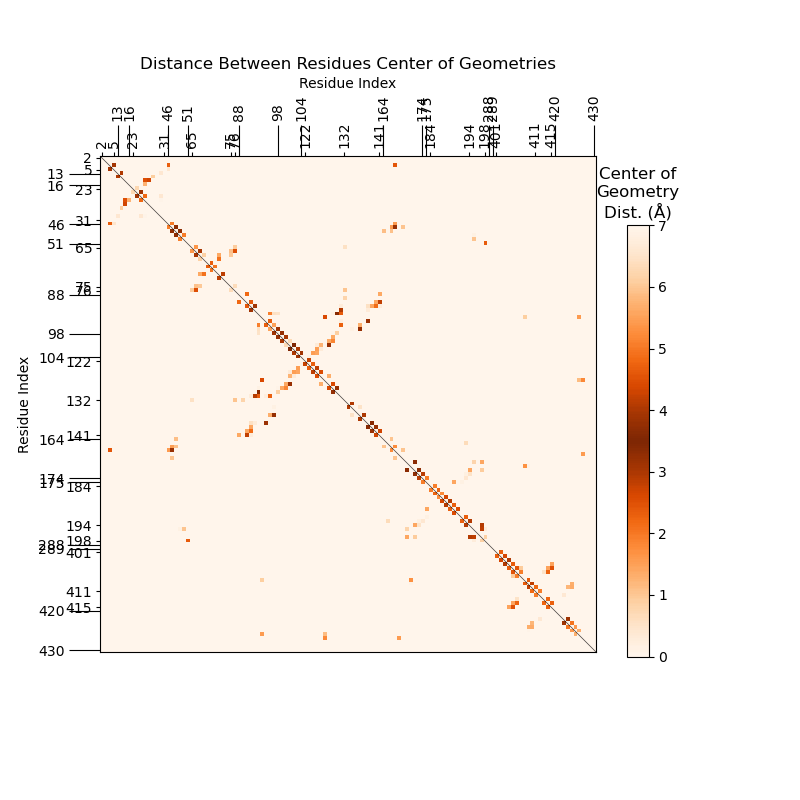
This will output the average SSF in minutes, as opposed to the hours it took to calculate
the -i 5JUP_N2_tGGG_aCCU_+1GCU_data.txt.gz input data.
Combined SSFs#
The line over x=y indicates that the SSF is symmetrical. This makes sense,
as the distance from residue i to j is the same as from j to i.
We can remove this redundancy by adding another trajectory’s SSF in the bottom left
half, for ease of comparison:
[user]$ stacker -s ssf \
-trj 5JUP_N2_tGGG_aCCU_+1GCU_nowat_2fpns.mdcrd \
-top 5JUP_N2_tGGG_aCCU_+1GCU_nowat.prmtop \
-r 2-5,13-16,23-31,46-51,65-76,88-104,122-141,164-175,184-198,288-289,401-415,420-430 \
-i 5JUP_N2_tGGG_aCCU_+1GCU_data.txt.gz \
-B 5JUP_N2_tGGG_aCCU_+1CGU_data.txt.gz
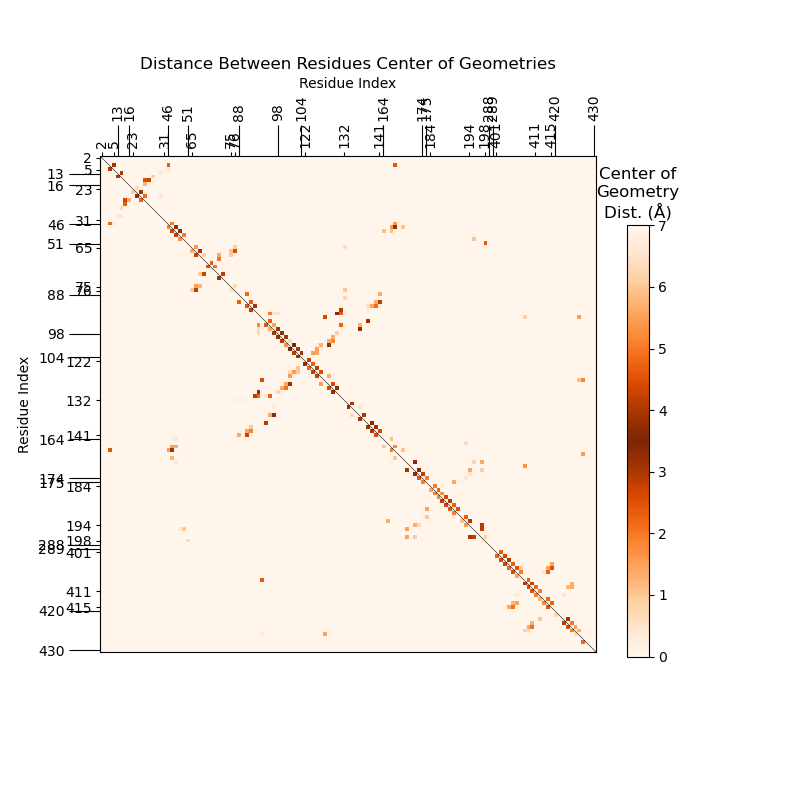
To get the residue pairs whose stacking changed the most between the two SSFs, see “Get Stacking Changes”.
Customizing SSFs#
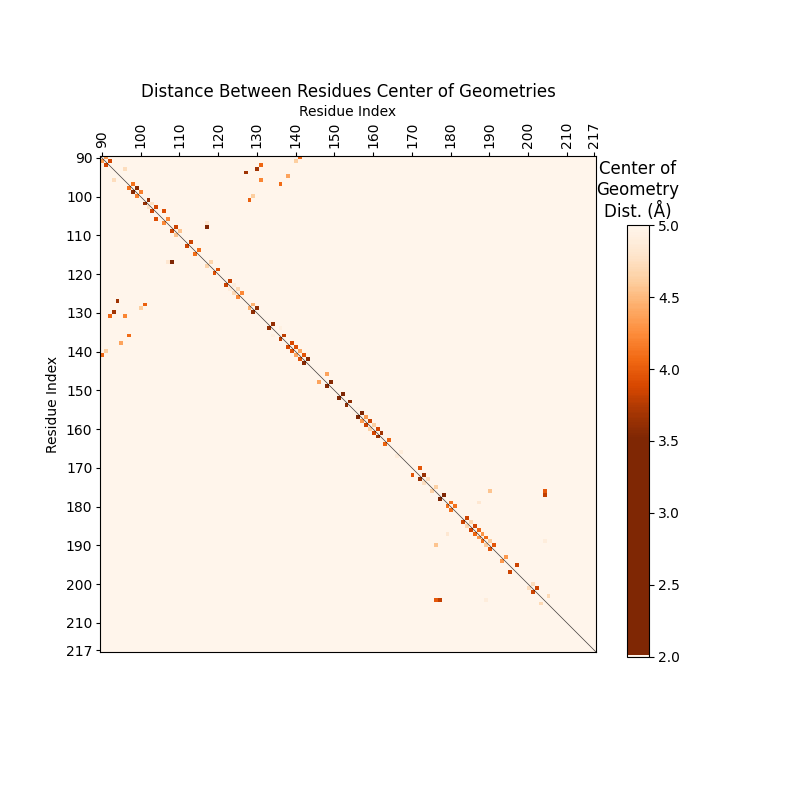
We can customize SSFs to color stacking events more stringently:
-l, --limits LIMITS: limits of the color scale, default = (0,7). Helps limit the pairings
visualized as pi-stacking.
- -y, --scale_style SCALE_STYLE : style of color scale. {bellcurve, gradient}
The SSFs we’ve displayed so far used the default limits = (0,7) and scale_style = bellcurve.
The scale_style parameter can help label rare <3.5Å stacking events as stacking rather than
the lighter color caused by bellcurve. Sterics generally prevent nucleotides from anything <3.4Å,
so this scale generally changes nothing and is a matter of personal preference.
[user]$ stacker -s ssf \
-trj first10_5JUP_N2_tUAG_aCUA_+1GCU_nowat.mdcrd \
-top 5JUP_N2_tUAG_aCUA_+1GCU_nowat.prmtop \
-r 90-215 \
-fl 1-10 \
-t 10 \
-l 2,5 \
-y gradient
Get Top Stacking Events#
stacker -s stack_events gives the top pi-stacking events (ie. the residue
pairs with COG distance closest to 3.5Å) for a given trajectory. A few important
flags:
- -i, --input INPUT_FILE : A .txt or .txt.gz containing per-frame SSF
data created by stacker -s ssf -d OUTFILE.
- -o, --output OUTPUT_FILE : spreadsheet output goes to a .txt file. Used for future analysis.
- -n, --n_events N_EVENTS : show top N_EVENTS stacking events. We use -n -1, which
outputs all residue pairs and their COG distance.
- -j, --include_adjacent : Include adjacent residues in the printed output. These
are much more likely to be stacking, you may be interested in only non-consecutive stacking residues.
[user]$ stacker -s stack_events \
-trj 5JUP_N2_tGGG_aCCU_+1GCU_nowat_2fpns.mdcrd \
-top TOPOLOGIES/5JUP_N2_tGGG_aCCU_+1GCU_nowat.prmtop \
-r 2-5,13-16,23-31,46-51,65-76,88-104,122-141,164-175,184-198,288-289,401-415,420-430 \
-i 5JUP_N2_tGGG_aCCU_+1GCU_data.txt.gz \
-o stack_events.GCU.txt \
-n -1 \
-j
WARNING: Residue Indices are expected to be 1-indexed
Reading trajectory...
Reading topology...
Filtering trajectory...
WARNING: Output filtered traj atom, residue, and chain indices are zero-indexed
The stacking events are outputted to stack_events.GCU.txt. We can look at this file
to find the residue pairs closest to 3.5Å apart—those that are pi-stacking.
[user]$ head stack_events.GCU.txt
Res1 Res2 Avg_Dist
101 102 3.55
139 138 3.61
130 93 3.63
172 170 3.68
172 173 3.70
48 49 3.71
47 48 3.72
129 130 3.76
136 97 3.78
Get Stacking Changes#
Once we have used stacker -s stack_events to get the top stacking events
for a single trajectory, we can run stacker -s compare to get the residue
pairs that changed the most between two different trajectories. These trajectories
must be comparable (ie. have the same number of residues). Generally, they should
be the same structure with very minor tweaks. The two trajectories used below are
the same ribosome PDB with one codon of the mRNA changed:
[user]$ stacker -s compare \
-A stack_events.GCU.txt \
-B stack_events.CGU.txt \
-SA _tGGG_aCCU_+1GCU \
-SB _tGGG_aCCU_+1CGU
Res1 Res2 Avg_Dist_tGGG_aCCU_+1GCU Avg_Dist_tGGG_aCCU_+1CGU Discrepancy
47 48 3.72 4.30 0.58
48 49 3.71 4.20 0.49
122 123 4.23 3.75 0.48
66 67 3.98 4.31 0.33
133 134 4.07 3.75 0.32
91 92 3.91 4.19 0.28
95 138 3.84 4.11 0.27
170 172 3.68 3.95 0.27
422 423 3.87 4.11 0.24
138 139 3.61 3.82 0.21
The output shows the average stacking distance between the residues in one trajectory compared to the other, and lists them by the residue pairs whose stacking distance changed the most.